Gene Editing Breakthrough: Multiplex Genome Editing for Complex Diseases
The new MOBE technology enables precise genome editing, opening new possibilities for modeling and understanding complex diseases like heart disease and schizophrenia

[Sciencephoto/Canva]
The human genome, a sprawling code composed of around three billion base pairs, reveals that humans are 99.6% genetically identical. The remaining 0.4%, a seemingly trivial variance, accounts for the vast tapestry of human differences. This fractional divergence holds crucial insights into complex health issues, from heart disease to neurodegenerative disorders like schizophrenia.
Yet, the challenge of modeling or correcting mutations in live cells remains daunting, particularly when attempting to install multiple point mutations simultaneously—a process known as multiplexing. Enter the innovative researchers at the University of California, San Diego, who have developed a groundbreaking set of tools named multiplexed orthogonal base editors (MOBEs). These tools, the brainchild of Assistant Professor of Chemistry and Biochemistry Alexis Komor’s lab, promise to revolutionize genome editing by enabling the simultaneous installation of multiple point mutations. Their findings, detailed in Nature Biotechnology, herald a new era in genetic research.
Komor's team fixated on genomes differing by a single letter in the DNA sequence (C (cytosine), T (thymine), G (guanine), and A (adenosine)), which are the fundamental bases of genetic material. In the grand narrative of human genetics, a single nucleotide variant (SNV) or point mutation could mean the difference between health and disease. Each individual carries about 4-5 million variants, some benign, others potentially deleterious, and often, it is a specific constellation of these variants that underpins disease.
Deciphering the genome's role in disease is complicated by the sheer volume of possible variations. For example, researchers seeking the genetic roots of heart disease must sift through the myriad differences in the genomes of affected individuals, a task akin to finding a needle in a genomic haystack.
“There is a problem interpreting genetic variants. In fact, most variants that are identified are unclassified clinically, so we don't even know if they’re pathogenic or benign,” stated Quinn T. Cowan, a recent PhD graduate from UCSD’s Department of Chemistry and Biochemistry and the paper's first author. “Our goal was to make a tool that can be used in disease modeling by installing multiple variants in a controlled laboratory setting where they can be studied further.”
To appreciate the significance of MOBEs, one must first understand the limitations of CRISPR-Cas9, the traditional gene-editing tool. CRISPR-Cas9 operates by using a guide RNA to direct the Cas9 enzyme to a specific genomic location, where it cuts both strands of DNA, creating a complete break. While effective, these double-stranded breaks can be toxic to cells and lead to unwanted insertions or deletions (indels) during the repair process. Multiplexing with CRISPR-Cas9 amplifies these risks.
In contrast, Komor's lab employs a base-editing technique, which induces a chemical change in the DNA, modifying just one letter at a time (such as converting a C to a T or an A to a G). This method is more meticulous and less damaging to cells and eschews the blunt-force approach of CRISPR-Cas9.
By deploying multiple base editors simultaneously, such as changing a C to T at one site and an A to G at another, researchers can more accurately model polygenic diseases—those caused by multiple genetic variants. However, previous attempts at such multiplexing were hampered by guide RNA "crosstalk," where base editors would make unintended changes.
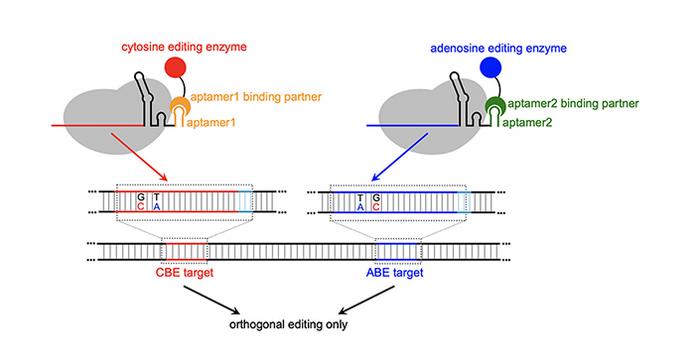
Cowan’s MOBEs employ RNA structures known as aptamers—tiny RNA loops that bind to specific proteins—to direct base-modifying enzymes to precise genomic locations. This innovation enables efficient, simultaneous editing of multiple sites with minimal crosstalk.
This pioneering approach marks the first use of aptamers to recruit adenosine base editors (ABEs) alongside cytosine base editors (CBEs) in an orthogonal pattern, forming the basis of MOBEs. The results are striking: without MOBEs, crosstalk between CBE and ABE occurs up to 30% of the time; with MOBEs, crosstalk plummets to under 5% while achieving a 30% conversion efficiency of the desired base changes.
Their study, a proof of concept, tested the MOBE system’s feasibility and secured a provisional patent. To further validate their tools, the team conducted case studies on real diseases, such as Kallmann syndrome, a rare hormonal disorder. Their experiments demonstrated that MOBE systems could effectively edit relevant cell lines for certain polygenic diseases.
“We’re in the process of putting the plasmids up on AddGene so anyone can freely access them. Our hope is that other researchers will use the MOBEs to model genetic diseases, learn how they manifest, and then hopefully create effective therapies,” stated Cowan.
With MOBEs, the frontier of genetic research broadens, promising not just to understand human disease but also to pioneer new therapies that may one day cure it.
Read More





Newletter & More









SynBioBeta
Join the innovators shaping the future with SynBio + AI. From health to ag, materials & more—be part of the revolution.



